


Pubblicata su the Lancet una review condotta da ricercatori italiani che ha fatto il punto sulla malattia di Behcet, una rara condizione clinica infiammatoria cronica multisistemica

E’ stata recentemente pubblicata su the Lancet una review condotta da ricercatori italiani del Dipartimento di Medicina Sperimentale e Clinica dell’Università di Firenze, che ha fatto il punto sulla malattia di Behcet, una rara condizione clinica infiammatoria cronica multisistemica.
Epidemiologia e patogenesi
La sindrome di Behçet è una malattia infiammatoria rara e multisistemica, caratterizzata dalla presenza di diverse manifestazioni cliniche diverse che presentano fasi imprevedibili di recidiva e remissione. Può insorgere a qualsiasi età, anche se la sua l’insorgenza presenta un picco nella terza decade di vita ed è rara nei bambini e nelle persone di età pari o superiore ai 50 anni. La prevalenza e la gravità della malattia sono generalmente più elevate negli individui di sesso maschile, anche se in alcuni Paesi è stata segnalata una prevalenza e una gravità maggiore nel sesso femminile.
La prevalenza globale della sindrome di Behçet è di 10,3 per 100.000 persone (IC95%: 6,1-17,7) per 100.000 persone, con un’elevata variabilità geografica, anche in termini di manifestazioni della malattia.
La sindrome di Behçet è stata anche definita come la malattia della “Via della Seta” per la sua prevalenza elevata nei Paesi eurasiatici, tra i 30°N e i 45°N di latitudine, che si trovano, appunto, lungo la cosiddetta “Via della Seta”.
È endemica in Medio Oriente, Asia orientale, e nel bacino del Mediterraneo, con un’elevata prevalenza in Turchia, Iran, Arabia Saudita, Iraq, Cina e Giappone. Negli ultimi 20 anni, però, è aumentata la prevalenza anche nei Paesi non endemici (tra cui l’Europa), decenni, l’incidenza della malattia è aumentata a causa delle migrazioni.
L’etiopatogenesi della sindrome è multifattoriale e, per quanto ancora oggi non del tutto compresa, sostenuta dalla relazione tra agenti infettivi e microbiota, genetica ed epigenetica, disfunzione del sistema immunitario e altri fattori ambientali.
Diagnosi
Ad oggi non esistono ancora biomarcatori validati o alcune caratteristiche istologiche in grado di identificare in maniera univoca la sindrome di Behcet. La diagnosi, pertanto, poggia sulla valutazione dei segni e dei sintomi clinici.
Allo stato attuale esistono più di 15 set di criteri per la classificazione e la diagnosi di Behcet: di questi, tre sono comunemente utilizzati nella pratica clinica e negli studi epidemiologici.
Manifestazioni cliniche
La diagnosi di malattia si applica a gruppi eterogenei di pazienti con differenti segni e sintomi, che possono presentarsi singolarmente i coesistere nella stessa fase temporale o in fasi differenti di malattia.
Lo spettro di manifestazioni cliniche è eterogeneo e comprende manifestazioni mucocutanee, articolari, oculari, vascolari, neurologiche e gastrointestinali che possono presentarsi con un decorso recidivante e remittente.
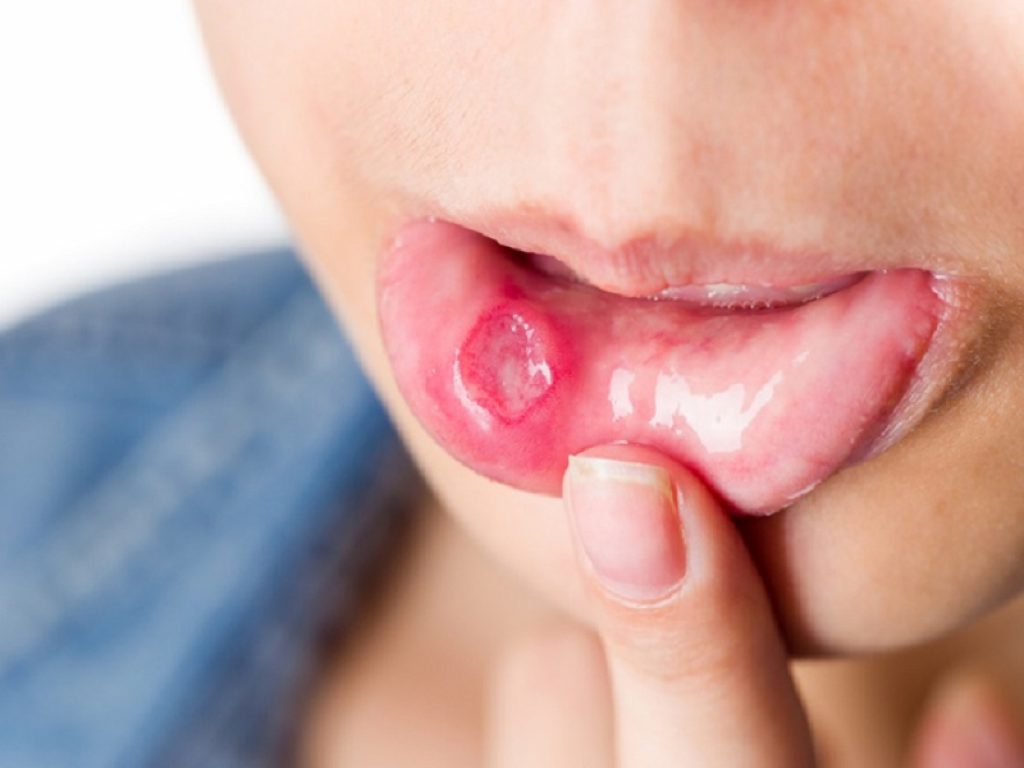
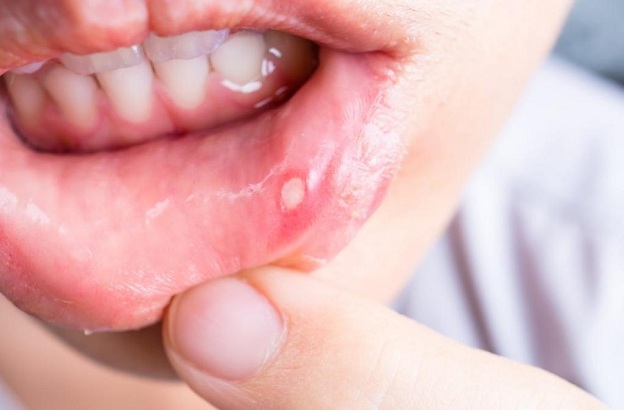
Le ulcere orali e genitali ricorrenti rappresentano le caratteristiche di più frequente riscontro della sindrome di Behcet, con una prevalenza fino al 95% nei pazienti di ambo i sessi.
Le lesioni cutanee iniziano spesso dopo l’adolescenza, colpiscono circa l’85% dei pazienti, e comprendono principalmente lesioni acneiche o papulopustolose su viso, parte superiore del torace, collo, spalle ed estremità, collo, spalle ed estremità.
L’interessamento articolare è presente nel 50-80% dei pazienti di ambo i sessi, con una distribuzione geografica variabile; le manifestazioni tipiche dell’interessamento articolare sono la monoartrite asimmetrica ricorrente, l’oligoartrite o le artralgie, di solito agli arti inferiori.
Le lesioni oculari possono essere rilevate in un paziente su due (prevalentemente di sesso maschile) e possono essere anteriori, intermedie, posteriori o da panuveite.
L’interessamento oculare inizia solitamente intorno alla fine della terza decade di vita e durante i primi 2-3 anni dalla diagnosi, rappresentando il primo segno di malattia di Behcet nel 20% dei pazienti.
Fino al 40% dei pazienti con sindrome di Behçet, per lo più di sesso maschile, sviluppano eventi vascolari, di solito nelle fasi iniziali della malattia. Le manifestazioni più comuni sono la trombosi venosa superficiale (TVS) e la trombosi venosa profonda (TVP), che in genere coinvolgono gli arti superiori o inferiori, con conseguente sindrome post-trombotica.
La trombosi in siti atipici può coinvolgere la vena cava inferiore e superiore, le vene epatiche con la sindrome di Budd-Chiari, la vena porta, i seni venosi cerebrali (CVS) o il ventricolo destro.
Il coinvolgimento arterioso è una caratteristica frequente e specifica della sindrome di Behçet, con aneurismi che coinvolgono i distretti periferico, viscerale e polmonare.
L’interessamento neurologico, noto anche come sindrome di neuro-Behcet, colpisce circa il 5% dei pazienti (per lo più di sesso maschile) e si manifesta di solito 5 anni dopo l’esordio della sindrome di Behçet.
Nell’80-90% dei pazienti è coinvolta la regione parenchimale, compreso il tronco encefalico, la giunzione telencefalica e diencefalica e i gangli della base, mentre il midollo spinale è meno frequentemente coinvolto.
Passando alle manifestazioni gastrointestinali, queste hanno una prevalenza simile nei pazienti con Behcet di ambo i sessi e compaiono di solo 5-10 anni dopo l’insorgenza delle ulcere orali.
I sintomi possono variare da una condizione asintomatica a lieve disagio addominale o a dolore grave, con ulcere localizzat principalmente nella regione ileocaecale terminale e, più raramente, nelle aree perianale e rettale.7
Nei casi più gravi si possono avere complicanze, come emorragie o perforazioni.
Tra le manifestazioni aggiuntive rare della sindrome di Behçet. Abbiamo l’interessamento otologico (che include perdita dell’udito e disturbi dell’andatura), anomalie del parenchima polmonare, coinvolgimento cardiaco e aterosclerosi, e problemi a carico della vescica urinaria.
Terapia farmacologica
Il trattamento della sindrome di Behçet si basa sull’impiego di diversi farmaci e varia in base all’interessamento di malattia.
Nello specifico:
– per il coinvolgimento mucocutaneo, si può partire con un trattamento con glucocorticoidi topici o sistemici a basso dosaggio. La colchicina, invece, rappresenta il trattamento di prima scelta per la prevenzione delle recidive.
Tra i farmaci di seconda linea raccomandati per i pazienti con sindrome di Behçet resistente e refrattaria abbiamo apremilast, azatioprina, i farmaci anti-TNF, talidomide e interferone alfa. Naturalmente, il rischio teratogeno associato alla talidomide deve essere sempre tenuto in considerazione.
Nei pazienti che sono resistenti, refrattari o intolleranti agli anti-TNF, si può ricorrere a farmaci alternativi promettenti, quali ustekinumab, secukinumab, gli inibitori di IL-1 e il mofetil micofenolato.
– per il coinvolgimento articolare, si dovrebbe partire inizialmente con un trattamento con colchicina. Se si verifica un attacco acuto monoarticolare, si può prendere in considerazione il ricorso alla terapia infiltrativa con glucocorticoidi, oppure con FANS o glucocorticoidi sistemici per un breve periodo di tempo.
Per i pazienti con manifestazioni ricorrenti o croniche, si raccomandano, come immunosoppressori di seconda linea, azatioprina, i farmaci anti-TNF e l’interferone alfa.
Tra le opzioni possibili di terza linea, invece, abbiamo secukinumab, apremilast, anakinra e tofacitinib.
– per il coinvolgimento oculare, i glucocorticoidi topici insieme ai farmaci cicloplegici o ipotonici rappresentano il trattamento di prima linea nell’uveite anteriore isolata.
Dovrebbe essere preso in considerazione, nei pazienti con fattori prognostici sfavorevoli (es. giovane età, sesso maschile, esordio precoce di malattia), l’impiego di immunosoppressori sistemici (azatioprina in particolare).
Alcune evidenze suffragano l’impiego dei farmaci anti-TNF nel prevenire le recidive di uveite anteriore legata al Behcet.
Anche il trattamento dell’uveite posteriore e della panuveite si basa, in primis, sull’impiego di farmaci immunosoppressori tradizionali per via sistemica (azatioprina o ciclosporina) o biologici (come l’interferone alfa i farmaci anti-TNF – infliximab o adalimumab in particolare). Inoltre, possono essere utilizzati glucocorticoidi ad alto dosaggio in combinazione con questi immunosoppressori.
Nei pazienti con coinvolgimento unilaterale acuto, si possono aggiungere iniezioni intravitreali di glucocorticoidi al trattamento sistemico.
Infine, nei pazienti refrattari, si suggerisce l’impiego di farmaci anti-IL-1 e di tocilizumab alla luce di alcune evidenze presenti.
– per il coinvolgimento vascolare, il trattamento varia a seconda del distretto coinvolto e del tipo di evento.
Gli immunosoppressori sono la pietra miliare del trattamento per gli eventi venosi e arteriosi, mentre il ruolo degli anticoagulanti è dibattuto. Nei pazienti con interessamento venoso di siti tipici, sono da considerare, come trattamento principale, i glucoorticoidi e gli immunosoppressori tradizionali (soprattutto azatioprina, ciclosporina e ciclofosfamide).
Nei pazienti con trombosi venosa refrattaria, si raccomanda l’impiego di farmaci anti-TNF, con o senza DMARDcs, o interferone alfa (in casi selezionati), anche in combinazione con anticoagulanti.
Nei pazienti con malattia multirefrattaria, stanno emergendo nuove evidenze a favore dell’impiego di farmaci alternativi, come il baricitinib.
L’associazione tra anticoagulanti e immunosoppressori (preferibilmente ciclofosfamide o anti-TNF) è raccomandata in caso di trombosi estese delle vene più grandi, in particolare della vena cava, e nei pazienti con trombosi dei seni venosi cererbrali (CVS) o con trombi intracardiaci.
Per quanto riguarda il coinvolgimento arterioso, i pazienti che presentano aneurismi polmonari o aortici devono essere trattati con glucocorticoidi a dosaggio elevato e ciclofosfamide o con farmaci anti-TNF (in particolare infliximab).
Dovrebbe essere preso in considerazione l’impiego di corticosteroidi e immunosoppressori (es: azatioprina) per i piccoli aneurismi, mentre l’azatioprina può essere utilizzata anche per il mantenimento della remissione.
– per il coinvolgimento parenchimale del sistema nervoso centrale (CNS), Il controllo iniziale degli attacchi neurologici acuti prevede l’impiego di glucocorticoidi a dosaggio elevato (tipicamente iniziando con 1 mg/kg al giorno per via orale con o senza un pulse endovena di 500-1000 mg/die, per 3-7 giorni consecutivi), seguiti da una lenta riduzione della loro posologia insieme all’impiego di farmaci immunosoppressori.
L’azatioprina rappresenta, di solito, il trattamento di prima linea per le forme moderate; in pazienti con coinvolgimento grave, persistente o recidivante, invece, deve essere iniziato il prima possibile un trattamento con farmaci anti-TNF o con ciclofosfamide.
Può essere preso in considerazione l’impiego di metotressato per il trattamento delle forme croniche progressive, mentre il mofetil micofenolato può essere un’opzione di trattamento per i pazienti con coinvolgimento di organi importanti che son intolleranti all’azatioprina o che sono in trattamento con warfarin.
Inoltre, alcune osservazioni supportano l’impiego di tocilizumab.
– per il coinvolgimento gastrointestinale, i pazienti devono essere inizialmente trattati, nei casi lievi, con derivati 5-aminosalicilati. Nei casi di gravità moderata-grave, va presa in considerazione, invece, l’assunzione di azatioprima, riservando i glucocorticoidi a dosi elevate per via orale o endovena nei casi più gravi, pur permanendo dubbi sul profilo rischio-beneficio.
Nei pazienti con gravi manifestazioni enteriche scarsamente controllate dall’azatioprina, invece, dovrebbe essere presa in considerazione l’assunzione di anti-TNF (infliximab o adalimumab in particolare, o talidomide (anche in combinazione con infliximab).
Da ultimo, alcune evidenze suggeriscono anche la possibilità di somministrare metotressato, ciclosporina, tacrolimus e baricitinib nel trattamento della sindrome.
Bibliografia
Emmi G et al. Behcet’s syndrome. Lancet 2024
Leggi